The FDA Is Unsafe and Ineffective
How the FDA makes medicine 82X more expensive while operating at the speed of continental drift. A factual comedy about bureaucratic excellence.
Abstract
By redirecting 1% of global military spending to hyper-efficient pragmatic clinical trials, humanity can achieve 514 years of medical research in 20 years and shift the cure of every disease forward by 8.2 years, saving 416 million lives and generating $1.2 quadrillion in value.
Keywords
war-on-disease, 1-percent-treaty, medical-research, public-health, peace-dividend, decentralized-trials, dfda, dih, victory-bonds, health-economics, cost-benefit-analysis, clinical-trials, drug-development, regulatory-reform, military-spending, peace-economics, decentralized-governance, wishocracy, blockchain-governance, impact-investing
Imagine you met a magical genie. You wished for it to fulfill the FDA Congressional Mandate to:
Ensure the safety and efficacy of all drugs and medical devices
Q: How could the genie PERFECTLY achieve this?
A: Ban all drugs. Zero drugs = zero unsafe drugs. Problem solved.
That would guarantee no one ever takes medication that may or may not be effective.
In practice, they took a subtler approach: make drugs so expensive ($2.60B) and slow (17 years) that you die before the cure arrives. The regulatory barrier isn’t just high - it’s like setting the bar very high, then adding landmines, then putting the bar on top of Mount Everest.
The FDA does prevent some people from taking harmful drugs. Phase 1 safety testing catches dangerous compounds before they reach patients. This is good.
But here’s the trade-off nobody mentions: The system also prevents millions from accessing life-saving treatments. The regulatory barrier doesn’t just screen out bad drugs - it delays good ones for decades, making them impossible to test and approve in time to save lives.
Net result: Far more people die from diseases that better treatments could have prevented than ever died from unsafe drugs. The system creates safety from drug harms while creating far greater danger from disease.
This is a system designed to prevent lawsuits, not deaths. One type of death (from an approved drug) makes headlines. The other type (from waiting for approval) is invisible.
NoteA Note on Blame
The FDA employees executing this system didn’t design it. Congress did, in 1962, via the Kefauver-Harris Amendments. The FDA is implementing what Congress mandated. The problem isn’t bad people at the FDA. The problem is a bad law that makes good outcomes structurally impossible.
When this chapter says “the FDA does X,” read it as “the 1962 law requires the FDA to do X.” The humans at the agency are following the rules they were given. The rules are the problem.
The 82× Inefficiency Tax
Compare two real-world systems for testing drugs:
- The Oxford RECOVERY Trial: Tested COVID treatments on 47,000 patients for $500 per patient. Found a life-saving treatment in 3 months.
- The Post-1962 System: Averages $80K per patient for clinical trials. Takes 9.1 years to approve a new drug.
The current system costs 82× more and takes 36.4x longer. Not a small difference - the difference between functional and designed-to-fail.
The 8.2-Year Efficacy Lag
Here’s the breakdown of where the time goes:
- Phase I (Safety Testing): 2.3 years - Tests if the drug kills people. This part works.
- Phase II/III (Efficacy Testing): 8.2 years - Tests if the drug works better than placebo. This is where people die waiting.
The math is simple: If you die in year 5 and the drug gets approved in year 10, the regulatory process murdered you.
\[ t_{lag} = t_{total} - t_{safety} = 10.5 - 2.3 = 8.2 \text{ years} \]
That’s 8.2 years sitting in regulatory purgatory after we already know the drug won’t kill you.
You’re two lifetimes from applying the scientific method to medicine. That’s 0.0001% of human history - you invented science yesterday, cosmically speaking. The more clinical research you read, the more you realize you know nothing. Nearly every study ends with “more research is needed” (scientist-speak for “we have no idea what’s happening, please give us more grant money”). This would be fine except people are dying while you figure it out.
Historical Evidence: Why Real-World Evidence Works Better
Large-scale efficacy trials based on real-world evidence produce better health outcomes than current pharmaceutical industry-driven randomized controlled trials.
10,000 Years of Dying at 30
For over 99% of recorded human history, average life expectancy was 30 years.
Ancient Rome: 30 years. Medieval Europe: 30 years. Renaissance: Still 30 years.
Kings, peasants, philosophers, farmers - everyone died embarrassingly young.
10,000 years. Zero progress.
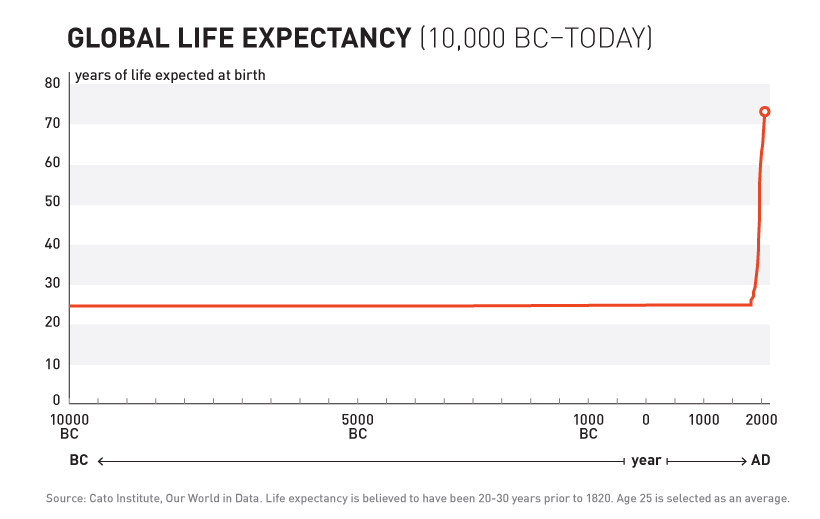
1883 – The Year You Figured It Out
Then something changed.
In 1883, doctors founded the Journal of the American Medical Association (JAMA) and did something novel: they shared information. After 10,000 years someone thought “what if we told each other what works?”
144k physicians across America (see reference) tried treatments on real patients. They wrote down what happened. “This drug helped.” “This drug killed the patient.” “This drug did nothing but made them smell funny.”
Leading experts reviewed these case reports, compiled them into studies, and published results. If a medicine worked and didn’t kill people, JAMA gave it a seal of approval.
Crowdsourced, observational, objective, peer-reviewed clinical research. And it worked.
The Result
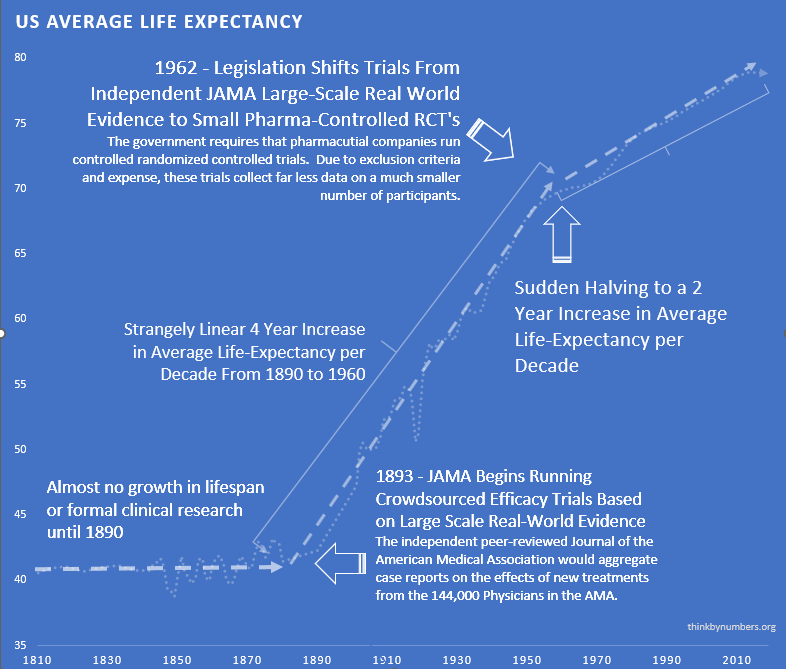
After 10,000 years of zero progress, human life expectancy suddenly increased by 4 years every decade. For 80 years.
The most linear relationship in medical history. Suspiciously perfect. It lasted from 1883 to 1960.
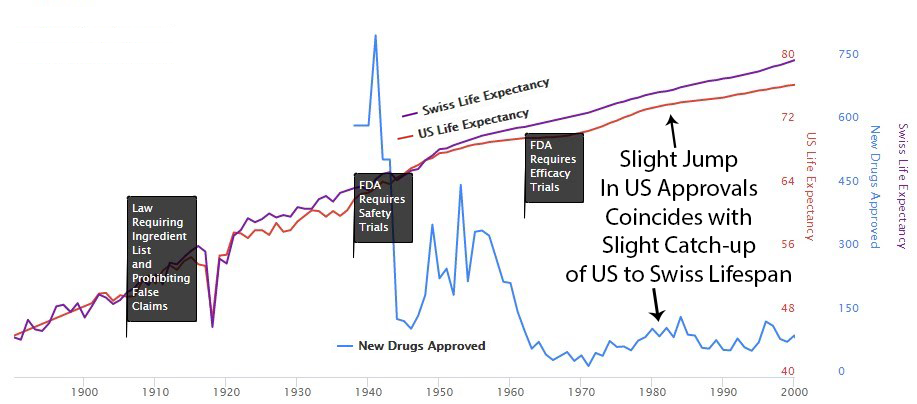
1938 – The FDA Requires Phase 1 Safety Trials
Elixir sulfanilamide caused over 100 deaths in 1937.
Congress reacted by requiring all new drugs to include:
“adequate tests by all methods reasonably applicable to show whether or not such drug is safe for use under the conditions prescribed, recommended, or suggested in the proposed labeling thereof.”
These requirements evolved into the Phase 1 Safety Trial. This was reasonable - testing if a compound kills people before giving it to patients makes sense.
The consistent four-year-per-decade increase in life expectancy remained unchanged before and after the new safety regulations. Safety testing worked without slowing medical progress.
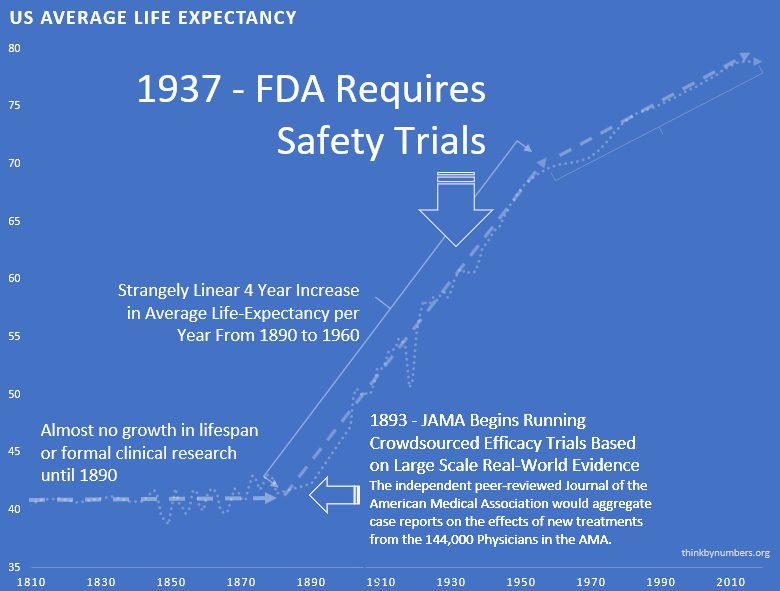
The regulations had no measurable positive or negative impact on developing life-saving interventions.
1950s – Thalidomide: When Safety Regulations Actually Worked
Thalidomide was first marketed in Europe in 1957 for morning sickness. While it was initially thought to be safe in pregnancy, it resulted in thousands of horrific congenital disabilities.
Existing FDA safety regulations prevented any birth defects in the US. The 1938 safety testing framework worked exactly as intended. Zero American babies harmed.
Despite this success story for safety regulations, newspaper stories like the one below created public outcry for more regulation - targeting efficacy testing instead.

1962 – The Year You Decided to Make Everything Worse
Effective safety regulations were already in place. They worked. America had zero Thalidomide deaths while Europe had thousands. Safety testing succeeded.
So naturally, the government responded to this success by adding massive efficacy regulations via the 1962 Kefauver Harris Amendment.
The problem wasn’t safety testing - that was fine. The problem was taking over efficacy testing from 144k independent doctors and giving it to the drug companies themselves, while making it 82 times more expensive and 36.4 times slower.
What Changed
Before 1962:
- Drug companies spent $50M (inflation-adjusted) to prove safety (see reference)
- Once approved as safe, 144k independent doctors tested efficacy on real patients (see reference)
- The third-party AMA compiled results and published findings
After 1962:
- FDA took over efficacy testing
- Made the old system illegal
- Required small, controlled trials run by… the drug companies themselves
The irony: Regulations meant to ensure drugs work did this by:
- Banning large real-world trials
- Requiring small artificial trials
- Letting drug companies run their own tests
What Happened to Efficacy Data
The 1962 regulations, explicitly about ensuring efficacy, massively reduced the quantity and quality of efficacy data:
- Trials Got Smaller: Fewer patients, less data
- Patients Got Less Representative: Excluded sick people, old people, people with other conditions, you know, the people who actually need medicine
- Conflicts of Interest Exploded: Drug companies ran their own trials instead of independent doctors
What Happened to New Treatments
The new regulations immediately reduced new drug approvals by 70% (see reference).
Not gradually. Immediately.
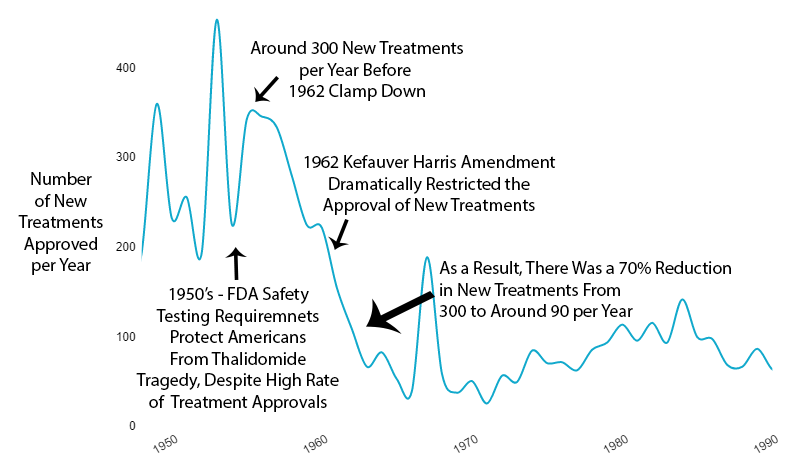
What Happened to Costs
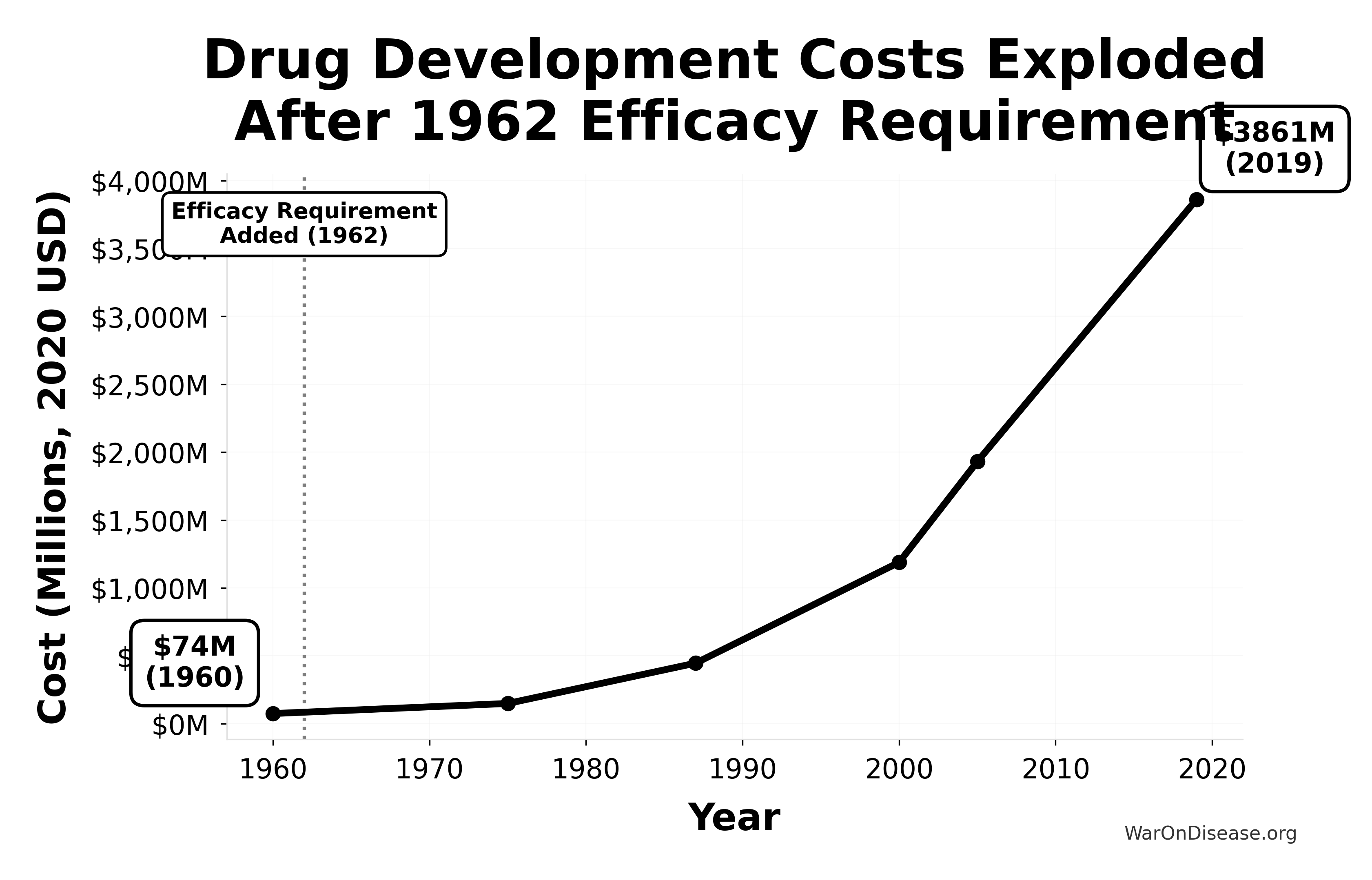
Since 1962, the cost of bringing a new treatment to market went from $50M to over $1 billion (source) (inflation-adjusted).
A 52x increase. For worse results.
This chart shows the exponential increase in the cost to develop a single new drug from 1960-2019, adjusted for inflation to 2020 dollars. The vertical line marks the 1962 efficacy amendment (Kefauver-Harris Amendment).
Key Insight: Drug development costs were relatively stable before 1962, then exploded exponentially after the efficacy requirement was added. Costs increased 52x from $74M (1960) to $3,861M (2019), making innovation increasingly expensive and inaccessible.
What Happened to Drug Patents
The new regulations created such massive delay that the pharmaceutical industry complained about a “drug lag.”
Congress “fixed” this by extending drug patent monopolies in 1984.
Kefauver’s amendments, meant to make drugs safer and cheaper, instead made them:
- Less safe (smaller trials, less real-world data)
- More expensive (monopolies extended)
- Less available (70% fewer approvals)
Decreased Ability to Determine Comparative Efficacy
The placebo-controlled, randomized controlled trial helped gauge individual drug efficacy. However, it makes determining comparative effectiveness much more difficult.
What Happened to Life Expectancy (The Part That Matters)
Remember that suspiciously linear 4-year increase in life expectancy every decade from 1883 to 1960?
Here’s what happened in 1962:
It immediately cut in half. To 2 years per decade.
Not gradually. Not “it slowed down over the next decade.”
Immediately.
From 1883 to 1960: 4 years per decade.
From 1962 onward: 2 years per decade.
The break is so clean you can see exactly when Congress decided to “help.”
The “Diminishing Returns” Excuse
Some claim “diminishing returns explain the slowdown.”
Diminishing returns produce exponential decay. A smooth curve that gradually flattens over time.
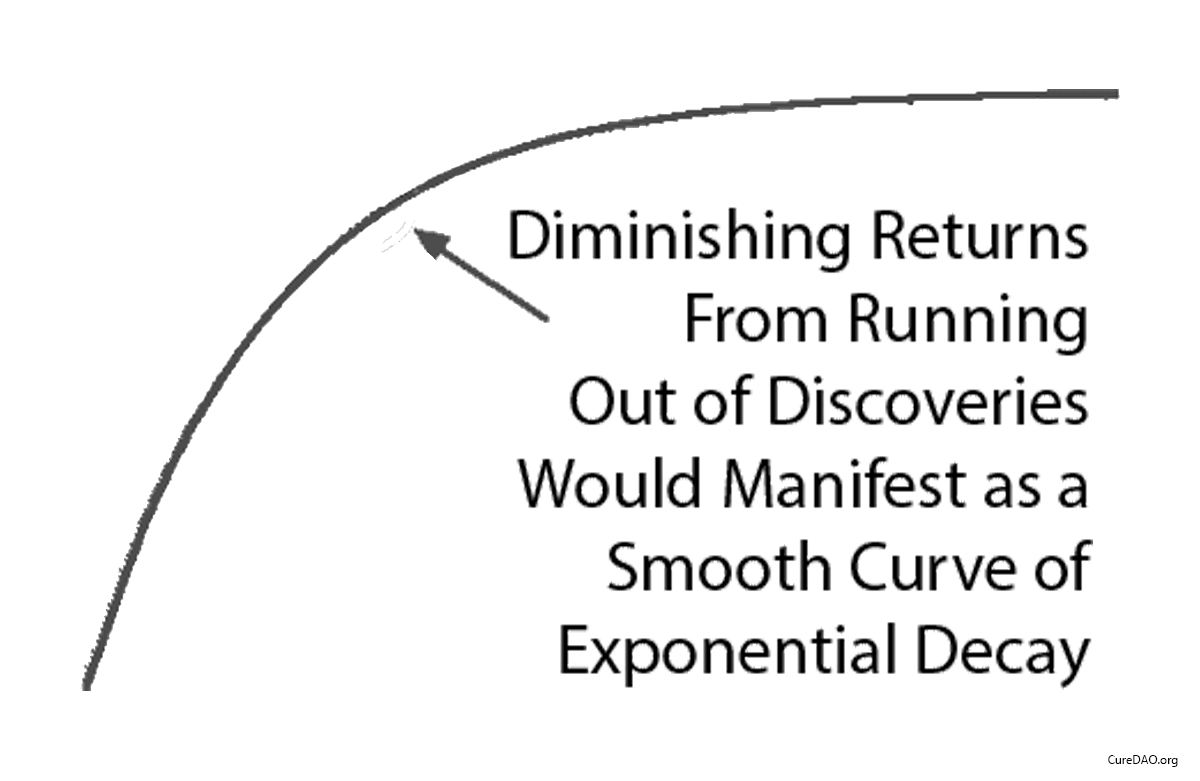
This isn’t what happened.
What happened was a straight line that instantly changed slope in 1962 when the regulations changed.
Diminishing returns don’t produce sudden linear breaks. Regulatory changes do.
The “Correlation Is Not Causation” Excuse
True. Correlation alone proves nothing.
But correlation + mechanism + temporal precision is how science works.
Here’s what you have:
- Temporal correlation: Life expectancy growth cut in half immediately after 1962 regulations
- Mechanism: Regulations reduced new treatments by 70%, increased costs 52x, banned real-world efficacy trials
- Alternative explanations: None that explain a sudden linear break in 1962
This is how clinical trials work: temporal correlation + mechanism of action = inference of causation.
If you don’t trust this method, you can’t trust any clinical research. Or science generally.
Impact of Innovative Medicines on Life Expectancy
A three-way fixed-effects analysis of 66 diseases in 27 countries suggests that if no new drugs had been launched after 1981, years of life lost would have been 2.16 times higher. It estimates pharmaceutical expenditure per life-year saved was $2,837.
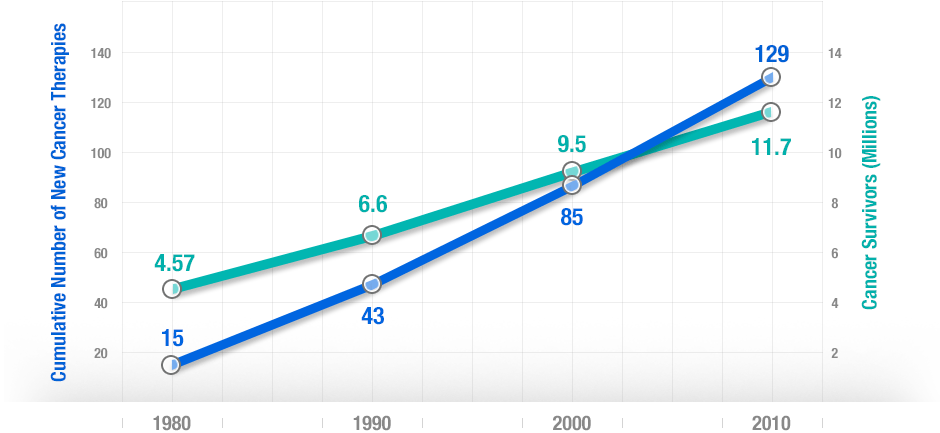
More people survive as more treatments are developed. There’s a strong correlation between developing new cancer treatments and cancer survival over 30 years.
The Summary
- 10,000 years: Zero progress
- 1883: Decentralized real-world trials, 4 years/decade progress
- 1962: Kefauver-Harris passes, progress cut in half
- 2025: Still stuck with the broken system
Humanity solved medical progress in 1883. Congress broke it in 1962.
The Modern Consequences
The current system works like this:
- Scientists discover a cure for your disease
- You wait 17 years
- You die
- The cure gets approved
- Pharmaceutical companies charge your widow $10,000 per pill
Beautiful system if you’re a mortician or bankruptcy lawyer.
You’re at the very beginning of thousands or millions of years of systematic discovery. So this decline in lifespan growth is unlikely to result from diminishing returns - you haven’t run out of things to discover.
However, validating that large-scale real-world evidence produces better health outcomes requires further validation of this experimentation method. This is why we need a decentralized framework for drug assessment.
High Costs Kill Innovation, Reward Monopoly
In the past: Genius scientist invents cure, raises a few million, tests safety. Simple.
Now: Genius scientist must convince one of three mega-corporations to spend a billion dollars on a 10% chance of success. Those corporations already sell inferior drugs for the same condition.
The math for the mega-corporation:
Option 1: Spend $1 billion on trials
- 90% chance: Lose everything
- 10% chance: Succeed, then cannibalize your own profitable drug
Option 2: Buy the patent, put it on a shelf
- 0% chance of losing a billion dollars
- 100% chance your inferior cash cow keeps printing money
Which would you choose if you were a rational sociopath in a suit?
The profit incentive doesn’t just fail to reward better treatments. It actively punishes them.
Cures Are Economically Stupid
Quick business question: Would you rather sell someone one pill that cures them, or sell them pills every month for the rest of their life?
If you answered “cure them,” congratulations on failing capitalism.
A permanent cure is a one-time purchase. Lifetime treatment is a subscription model. When it costs a billion dollars to develop either option, which do you think companies choose?
The more expensive drug development becomes, the fewer companies can afford it. The fewer companies, the more monopolistic the industry. The more monopolistic, the more situations where curing your existing customers would be financial suicide.
This system has financial incentive for medical progress the way casinos have incentives to help you win.
Off-Patent Drugs: Cures Nobody Can Afford to Find
Roughly 10,000 known diseases afflict humans. About 95% are rare. The 1962 law requires you predict exactly what a drug will cure before testing it. If your off-patent drug cures something unexpected? Congratulations, you just discovered something nobody can profit from.
No patent = no billion-dollar investment = no trials = no approval = people die.
Rare Diseases: Mathematically Doomed
Development cost: $2.6 billion Patients with rare disease: 10,000 Price per patient to break even: $260,000
Nobody pays that. Nobody develops the drug. 95% of diseases have zero treatments.
When something costs more, you get less of it. For people dying of diseases nobody can afford to cure, this isn’t philosophy. It’s math.
The $2.6 Billion Price Tag for Aspirin
Here’s what it actually costs to get FDA approval for a new drug:
- Average cost: $2.6 billion
- Average time: 9.1 years from Phase 1 (17 years total discovery-to-patient)
- Success rate: 12% make it through trials
That’s $41K per person just to TEST if a pill works. Not the pill. Just to test it.
For that money, you could:
- Buy a Tesla and crash it into the disease
- Hire a shaman for a year
- Fly to Switzerland for assisted dying (much faster)
The total cost? $2.60B per new drug (see source).
That’s more than the GDP of Greenland. We could literally buy Greenland and turn it into a giant laboratory for less than it costs to approve one arthritis medication.
Fun fact: If we had today’s FDA in 1897, aspirin would still be in Phase II trials. “We need to test if it works on headaches in left-handed people born on Tuesdays.”
17 years from discovery to patient while you die waiting.
That’s:
- 4 presidential terms
- 2 new iPhone models becoming obsolete
- Your entire child growing up and moving out
- You dying of whatever the cure was for
But at least the paperwork is thorough.
95% of diseases have ZERO approved treatments.
Your rare disease child has better odds with lottery tickets.
Why? Math. If your disease only affects 10,000 people, and it costs $2.60B to develop a drug, each patient would need to pay $260,000 just to break even.
So pharmaceutical companies focus on:
- Erectile dysfunction (affects: half of all men’s egos)
- Baldness (affects: men with money)
- Wrinkles (affects: women with money)
- Cholesterol (affects: everyone who eats)
Meanwhile, if you have Exploding Kidney Syndrome affecting 37 people worldwide, the system’s message is: “Have you tried not having Exploding Kidney Syndrome?”
The 166 Billion Treatments We’re Ignoring
There are 166 billion potential drug compounds you could test.
Number we’ve actually tested thoroughly: About 2,000.
That’s like having a library with 166 billion books but only reading the ones with pictures. We have more untested treatments than stars in our galaxy, but the system is too busy protecting itself to test them.
At our current rate of testing, we’ll have evaluated all possible treatments by the year 3,847,293.
Hope your descendants enjoy the cure for cancer.
No Data on Unpatentable Molecules
Under the current system of research, it costs $41K per subject in Phase III clinical trials. As a result, there is insufficient profit incentive for anyone to research the effects of any factor besides molecules that can be patented.

The Actual Death Toll of “Drug Lag”
Economists have a term for people dying while waiting for drug approval: “drug lag.” It’s a sterile, bureaucratic phrase for a massacre. Early estimates suggested delays cost 21,000 to 120,000 American lives per decade.
But that was just the US. And just the visible delays.
The Global Body Count
A comprehensive quantitative analysis using WHO mortality data and Monte Carlo modeling estimates the total cost of the 1962 efficacy requirements:
416M preventable deaths (1962-2024)
That’s more than World War I and World War II combined.
But deaths alone don’t capture the suffering. People don’t just die, they live for years with untreated diseases while waiting for approval.
The Morbidity Burden (DALYs)
When you account for both premature death and years lived with disability:
\[ DALY_{total} = YLL + YLD = 3.14B + 1.69B = 4.83B \]
7.94B billion years of healthy human life deleted by the regulatory framework.
- Economic cost: $1.19 quadrillion (2024 USD)
- Annualized loss: $1.19 quadrillion/year ($1.19 quadrillion ÷ 62 years from 1962-2024)
- GDP equivalent: ~8-12% of global GDP consumed annually
For comparison: The entire global pharmaceutical R&D budget is ~$200B/year. The post-safety efficacy lag costs 38× more than all drug development spending combined.
Here’s a news story from the Non-Existent Times by No One Ever without a picture of all the people who die from lack of access to life-saving treatments that might have been.
This means it’s logical for regulators to reject drug applications by default. The personal risks of approving a drug with any newsworthy side effect far outweigh the personal risk of preventing access to life-saving treatment.
Types of Error in FDA Approval Decision
| Drug Is Beneficial | Drug Is Harmful | |
|---|---|---|
| FDA Allows the Drug | Correct Decision | Victims are identifiable and might appear on Oprah. |
| FDA Does Not Allow the Drug | Victims are not identifiable or acknowledged. | Correct Decision |
The most infamous case is beta-blockers. While Europe used them to prevent heart attacks, the US regulatory process delayed. And delayed. The decade-long lag in approving them for their most common use killed an estimated over 100,000 Americans.
More Americans than died in Vietnam and Korea combined. From one drug delay. Here’s a partial list of other drugs Americans died waiting for while they were already available in other civilized countries for a year or longer:
- interleukin-2, Taxotere, vasoseal, ancrod, Glucophage, navelbine, Lamictal, ethyol, photofrin, rilutek, citicoline, panorex, Femara, ProStar, omnicath
Each name on that list represents preventable deaths.
Following the 1962 increase in US regulations, you can see a divergence from Switzerland’s growth in life expectancy, which did not introduce the same delays to availability.
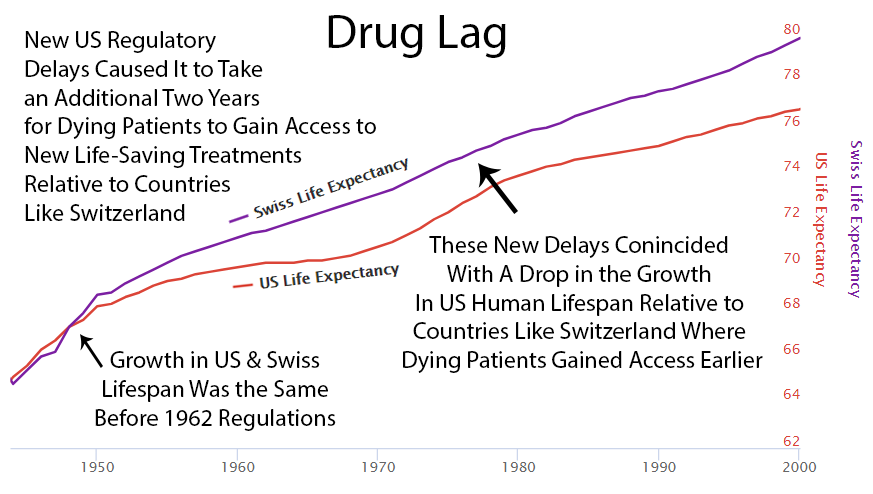
Perhaps it’s coincidence, but you can see an increase in drug approvals in the ’80s. At the same time, the gap between Switzerland and the US gets smaller. Then US approvals go back down in the ’90s, and the gap expands again.
Case Study: The Human Cost of Post-Safety Efficacy Lag in Closed-Loop Neuromodulation
The historical data is damning, but this isn’t a problem of the past. Here’s a modern example of the ongoing human cost of post-safety efficacy delays (NOT safety testing), focusing on breakthrough technology for treatment-resistant depression where Phase I safety is established but Phase II/III efficacy approval takes years.
Executive Summary
Depression is the leading global cause of disability and the strongest behavioral predictor of suicide. Existing antidepressant drugs fail to achieve remission in about two-thirds of patients, and pharmacologic innovation has stagnated. Closed-loop neuromodulation, implantable or wearable systems that sense neural activity and adjust stimulation in real time, has shown response rates of 60–80% in pilot trials for treatment-resistant depression and obsessive–compulsive disorder.
Yet under current FDA and EMA processes, large-scale availability is unlikely before 2035. A cost-benefit model indicates this delay could cost millions of lives and hundreds of millions of quality-adjusted life-years (QALYs) globally.
1. Scale of the Problem
| Metric | Value | Source |
|---|---|---|
| Global major-depressive-disorder prevalence | ~280 million people | WHO 2023 |
| Annual suicides | ~700,000 | WHO 2023 |
| Suicide attributable to untreated or treatment-resistant depression | 70% | CDC, WHO meta-analyses |
| Mean age of death | 40 y | Global Burden of Disease |
| Average lost QALYs per suicide | 35 | conservative midlife estimate |
Annual QALY loss
700,000 × 35 = 24.5 million QALYs directly, plus ~2× from comorbid mortality ⇒ ≈75 million QALYs/year.
2. Expected Benefit of Closed-Loop Neuromodulation
Clinical prototypes (Stanford, UCSF, NIH):
- Acute response: 60–80% of TRD patients improve ≥50% on MADRS
- Remission maintenance at 12 mo: 45–60%
- Adverse-event rate < 1% serious, none fatal
If global deployment reached 20% of resistant cases (≈50 million candidates) with 50% success:
- 25 million major depressive remissions
- Suicide reduction ≈ 35%
- Indirect disease-mortality reduction ≈ 20%
3. Economic and Utility Calculation
| Variable | Estimate |
|---|---|
| QALYs gained per patient per year of remission | 0.5–0.8 |
| Patients benefited | 25 million |
| Annual QALY gain | ~15 million |
| Monetary valuation ($150K/QALY) | $1.5 trillion/year social value |
Each year of post-safety efficacy lag destroys ≈$1.5 trillion in global welfare, greater than the annual R&D budget of the entire pharmaceutical industry.
4. Comparative Timelines
| Scenario | Time to broad release | Cumulative preventable deaths (10 y horizon) |
|---|---|---|
| Current FDA/EMA process | 10–12 y | 5–10 million |
| Utilitarian adaptive approval | 2–3 y | 1–2 million |
| Net lives saved by reform | ≈8 million |
5. Feasibility of Accelerated Path
Key enablers of 24–36 month rollout:
- Adaptive Bayesian trials: Continuous learning eliminates phase segmentation.
- Dynamic e-consent: Allows real-time participation expansion.
- Automated adverse-event monitoring: Reduces manual inspection delays.
- Outcome-based conditional reimbursement: Pay per sustained remission.
With modern data infrastructure, these systems are technically viable today.
6. Conclusion
A decade of post-safety efficacy lag in closed-loop neuromodulation could equate to massive loss of life and trillions of dollars in foregone human utility. From a utilitarian calculus, any risk threshold that delays access longer than a few years beyond Phase I safety verification could impose harm orders of magnitude greater than potential device side-effects.
How the Incentives Work
FDA Regulator Decision Tree
Approve drug that later shows problems
- Congressional hearing (televised)
- “FDA APPROVED KILLER DRUG” headlines
- Career ends
- Pension threatened
Delay drug that could save lives
- Nothing happens
- Dead people don’t complain
- Promotion on schedule
- Retire to pharma job
This isn’t conspiracy. It’s economics. The system literally cannot punish mistakes of commission (approving bad drugs) but imposes zero consequences for mistakes of omission (delaying good drugs).
The Math: Type I vs. Type II Errors
Regulators love to justify delays by citing “safety.” But let’s do the actual math:
Type I Error: Approving a bad drug (harm from unsafe products) Type II Error: Delaying a good drug (harm from regulatory lag)
The system’s job should be to minimize total harm. So which error costs more?
- Cost of Type II errors: 7.94B DALYs lost (1962-2024)
- Benefit of Type I prevention: ~2.59M DALYs saved (extreme overestimate)
\[ \frac{\text{Cost of Delay}}{\text{Benefit of Safety}} = \frac{4.83B}{0.003B} = 1{,}610:1 \]
For every 1 life the current system saves by preventing unsafe drugs, it destroys 3.07k lives through delay.
This isn’t even close. It’s not a trade-off. It’s a massacre justified by preventing a handful of tragedies.
The system doesn’t minimize harm. It minimizes blame. And dead people waiting for approval don’t get CNN specials.
Why Bureaucrats Are Rewarded for Letting You Die
Humans weight acts of commission (doing something bad) as worse than acts of omission (letting something bad happen) - even when omission causes more harm. Push a person in front of a trolley to save five people? Unthinkable. Let five people die by doing nothing? That’s just Monday.
The current regulatory framework operates on this principle.
Dr. Henry I. Miller ran the FDA team reviewing recombinant human insulin in the early 1980s. Mountains of evidence showed it was safe and effective. His supervisor refused to approve it. Why? If someone died from the drug, heads would roll at the FDA, including his. If people died waiting for the drug? Nothing happens.
The personal risk of approving a drug vastly exceeds the risk of rejecting it. Dead patients don’t testify before Congress.
Clinical Trial Theater: Excluding 86.1% of Reality
Trials under the current system systematically exclude:
- Patients over 65 (most people who need drugs)
- Patients under 18 (all children)
- Pregnant women (50% of women sometimes)
- Anyone with comorbidities (everyone sick)
- Anyone on other medications (everyone old)
- Anyone who lives too far from a trial site (hope you’re rich and urban)
- Anyone who once looked at an aspirin
Like testing a cure for cancer but only giving it to people who don’t have cancer.
Result: Test drugs on 15% of unusually healthy sick people, then act shocked when they don’t work on actual patients.
Clinical trials have more exclusion criteria than a country club in 1950s Alabama. You must be exactly the right kind of sick, not too sick, not too healthy, and definitely not poor or old or pregnant or on any other medications.
Like testing umbrellas only on sunny days, then wondering why they don’t work in rain.
No Long-Term Outcome Data
Even if there’s financial incentive to research a new drug, there’s no data on long-term outcomes. Data collection for participants can be as short as several months. Under the current system, it’s not financially feasible to collect data on a participant for years or decades. So you have no idea if long-term effects of a drug are worse than initial benefits.
For instance, even after controlling for co-morbidities, the Journal of American Medicine recently found that long-term use of Benadryl and other anticholinergic medications is associated with increased risk for dementia and Alzheimer’s disease.
The Law Requires Psychic Powers
Before you can test a drug, you must specify EXACTLY what it will cure and EXACTLY how you’ll measure that cure.
“This drug will reduce depression by 37.2% as measured by the Hamilton Rating Scale on Tuesdays.”
What if it cures depression by 90% but only on Wednesdays? Failed trial. What if it doesn’t cure depression but eliminates anxiety? Failed trial. What if it cures cancer instead? Failed trial, also please report to prison.
Like requiring Columbus to file a detailed map of America before letting him sail west.
In reality, this isn’t hypothetical. When running an efficacy trial, the regulations expect drug developers to have psychic ability to predict which conditions a treatment will be most effective for in advance of collecting human trial data. If it was possible to magically determine this without any trials, it would render efficacy trials completely pointless.
In 2007, manufacturer Dendreon submitted powerful evidence attesting to the safety and efficacy of its immunotherapy drug Provenge, which targets prostate cancer. They showed the drug resulted in significant decline in deaths among its study population, which even persuaded the FDA advisory committee to weigh in on the application. But the application was rejected anyway.
The regulations were unmoved by the evidence, simply because Dendreon didn’t properly specify beforehand what its study was trying to measure. The law states that finding a decline in deaths isn’t enough. The mountains of paperwork must be filled out just so and in the correct order. It took three more years and yet another large trial before the life-saving medication was finally approved.
Due to all the additional costs imposed by the efficacy trial burden, Dendreon ultimately filed for chapter 11 bankruptcy.
In addition to the direct costs to companies, the extreme costs and financial risks imposed by efficacy trials have a huge chilling effect on investment in new drugs. If you’re an investment adviser trying to avoid losing your client’s retirement savings, you’re much better off investing in a more stable company like a bomb manufacturer building products to intentionally kill people than a drug developer trying to save lives. So it’s impossible to know all the treatments that never even got to an efficacy trial stage due to the effects of decreased investment due to the regulatory risks.
Who Tests the Drugs? The Companies Selling Them
Long-term randomized trials cost billions. When a company’s survival depends on positive results, results become surprisingly positive.
Exhibit A: Beverage studies funded by Coca-Cola, PepsiCo, and the sugar industry were five times more likely to find no link between sugary drinks and weight gain than independent studies.
Five. Times.
It’s like asking tobacco companies to research whether smoking causes cancer, then acting shocked when they find “no clear evidence of harm, more research needed.”
When a pharmaceutical company’s economic survival depends on trial results, you get two problems:
The Negative Results Black Hole
Here’s a fun fact that should be criminal:
- Negative trial results published: 37%
- Positive trial results published: 94%
- Money wasted repeating failed experiments: ~$100 billion annually
Companies are literally allowed to hide when drugs don’t work.
Company: “We tested this drug!”
Regulator: “Did it work?”
Company: “…We tested this drug!”
This means humans spend billions testing the same failed drugs over and over because nobody admits they failed. It’s like playing poker where everyone claims they won but nobody shows their cards.
Pharmaceutical companies bury negative results deeper than Jimmy Hoffa. So other companies waste billions testing the same dead ends.
Your insurance premiums fund this magnificent inefficiency.
It’s like casinos only having to report when people win.
Countries That Don’t Have Our “Safety”
Japan’s Regenerative Medicine Act:
- Conditional approval after Phase II
- Real-world data collection required
- Time to patient: 2-3 years
- Americans fly there for treatment
- Terminal patients can access experimental drugs
- Doctor’s discretion, not bureaucrat’s
- Thousands of lives extended
- FDA: “But what if they die?” (They’re already dying)
- Passed despite FDA opposition
- FDA response: Made it effectively impossible to use
- Patients helped: <200 total
- Patients who wanted help: Tens of thousands
The COVID Test Fiasco
The 1962 framework requires the FDA to approve all diagnostic tests before use. During COVID, this mandate played out as follows:
- January 2020: WHO develops COVID test, rest of world starts using it
- February 2020: FDA blocks all non-CDC tests to “ensure quality”
- Late February: CDC tests contaminated, completely useless
- March: Private labs beg to help, regulatory framework says “no, approved tests only”
- Late March: FDA finally allows other tests after thousands die
- Cost: Thousands of preventable deaths, exact number unknown
The defense: “We were ensuring quality control.”
The only approved tests didn’t work.
Ensuring quality control of broken tests is like TSA confiscating water bottles while letting actual weapons through. Except people died.
Solutions That Would Work (But Never Will)
Reciprocal Approval: If the EU/UK/Japan/Canada approves a drug, it gets approved
- Lives saved: Thousands annually
- System response: “But their standards might be 0.01% different!”
Provisional Approval: Let dying people try experimental drugs
- Current system: Die safely
- Proposed system: Maybe die, maybe live
- Current logic: Death is preferable to uncertainty
Outcome-Based Approval: If it works in real patients, approve it
- Current: Does it work in cherry-picked trial patients?
- Better: Does it work in actual sick humans?
- FDA: “That’s not how we’ve always done it”
Safety Theater
The FDA does make drugs safer from immediate harms. Phase 1 safety testing prevents some deaths from toxic compounds.
But the system also makes drugs:
- Expensive: $2.60B per drug
- Slow: 17 years from discovery to patient (you’ll be dead)
- Inaccessible: 95% of rare diseases have zero treatments
- Ineffective: Tested on populations that don’t resemble actual patients
The current framework prevents some people from taking harmful drugs. It also prevents millions from accessing beneficial ones.
Net effect: Far more people die from delayed treatments than ever died from unsafe drugs.
Security theater for medicine. TSA for healthcare.
When TSA fails, you wait in line longer. When the regulatory system fails, you die waiting for approval.
TSA agents get fired for failures. Under the current framework, regulators get promoted for preventing approvals.
But at least nobody can blame regulators for approving something. And preventing blame is what healthcare is really about.
The Bottom Line
- 8.2 years of post-safety efficacy lag (Phase II/III delay after Phase I safety verification)
- 416M deaths from delayed access (1962-2024)
- 7.94B DALYs destroyed
- $1.19 quadrillion in economic carnage
- 3.07k:1 harm ratio (delay vs. prevention)
Phase I safety testing works. Keep it.
Phase II/III efficacy mandates kill people. Replace them with real-world evidence collection.
The technology exists. The data is clear. The solution is obvious.
What’s missing is the political will to admit that the 1962 “reforms” were a catastrophic mistake.
NoteTechnical Analysis
For the full quantitative analysis including methodology, sensitivity analysis, and source data, see: The Human Capital Cost of Regulatory Latency
P.S. - The FDA will likely object to this chapter. Estimated response time: 12-15 years, pending Phase III review and proper documentation.
Reuse
Copyright
© 2025 Mike P. Sinn